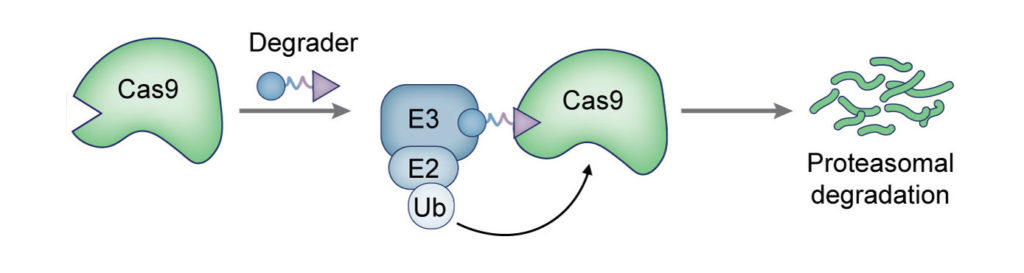
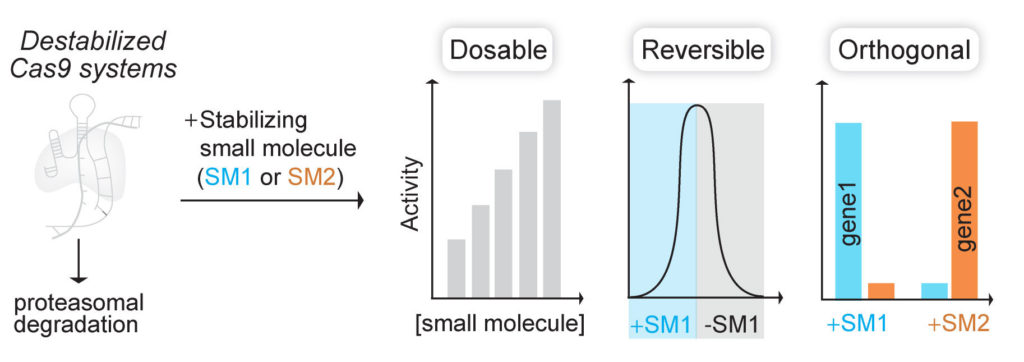
(Nat Chem Biol 2017). We fused Cas9 to destabilized domains that are largely unfolded and targeted, along with Cas9, for rapid proteasomal degradation. Addition of small molecule stabilizes these domains and rescues the fusion protein from degradation. By using this engineered Cas9 system, we were able to rapidly, reversibly, and dose-dependently control both genome editing and transcription technologies of Cas9.
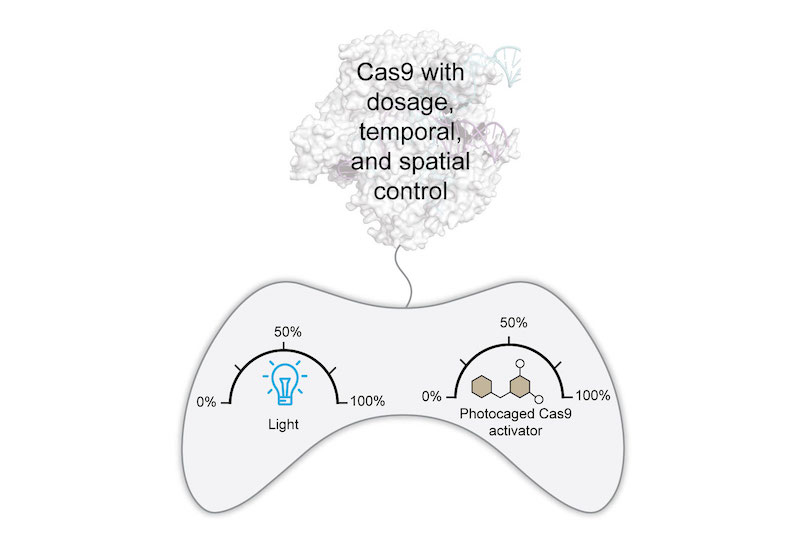
(Angew Chem Int Ed 2019). High-resolution, non-invasive, spatiotemporal control of Cas9 activity can be achieved using light. We photocaged our small-molecule activator to afford a singular system with precise dose and spatiotemporal control of Cas9 activity. This system is rapidly responsive, requires low light intensity and exposure times, and provides reversible and multi-wavelength control of Cas9.
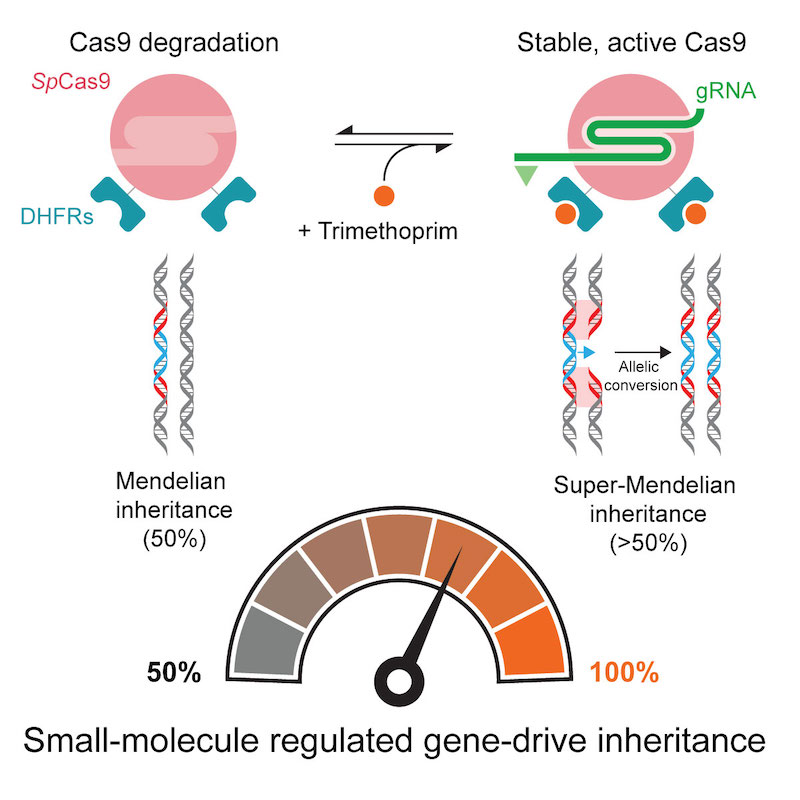
(Cell Reports 2020). CRISPR-based gene drives have the potential to fight vector-borne diseases or suppress crop pests because they can rapidly transmit important genes through a population. They do this by surpassing the 50% inheritance limit of Mendel’s law of independent assortment through their gene-editing technology that converts a wildtype allele to a gene-drive one. However, contemporary gene drives could spread through populations to permanently modify the genome of organisms, posing safety concerns that limit their use in both laboratory and field settings if left unchecked. We describe in Drosophila the first gene-drive system controlled by an engineered Cas9 and a synthetic, orally-available small molecule, which can be applied to field applications.
(Chemical Sciences 2019) We have developed a sensitive, high-throughput, cell-free method wherein Cas nuclease activity is amplified via an in vitro transcription reaction that produces a fluorescent RNA:small-molecule adduct.

(Cell 2019).
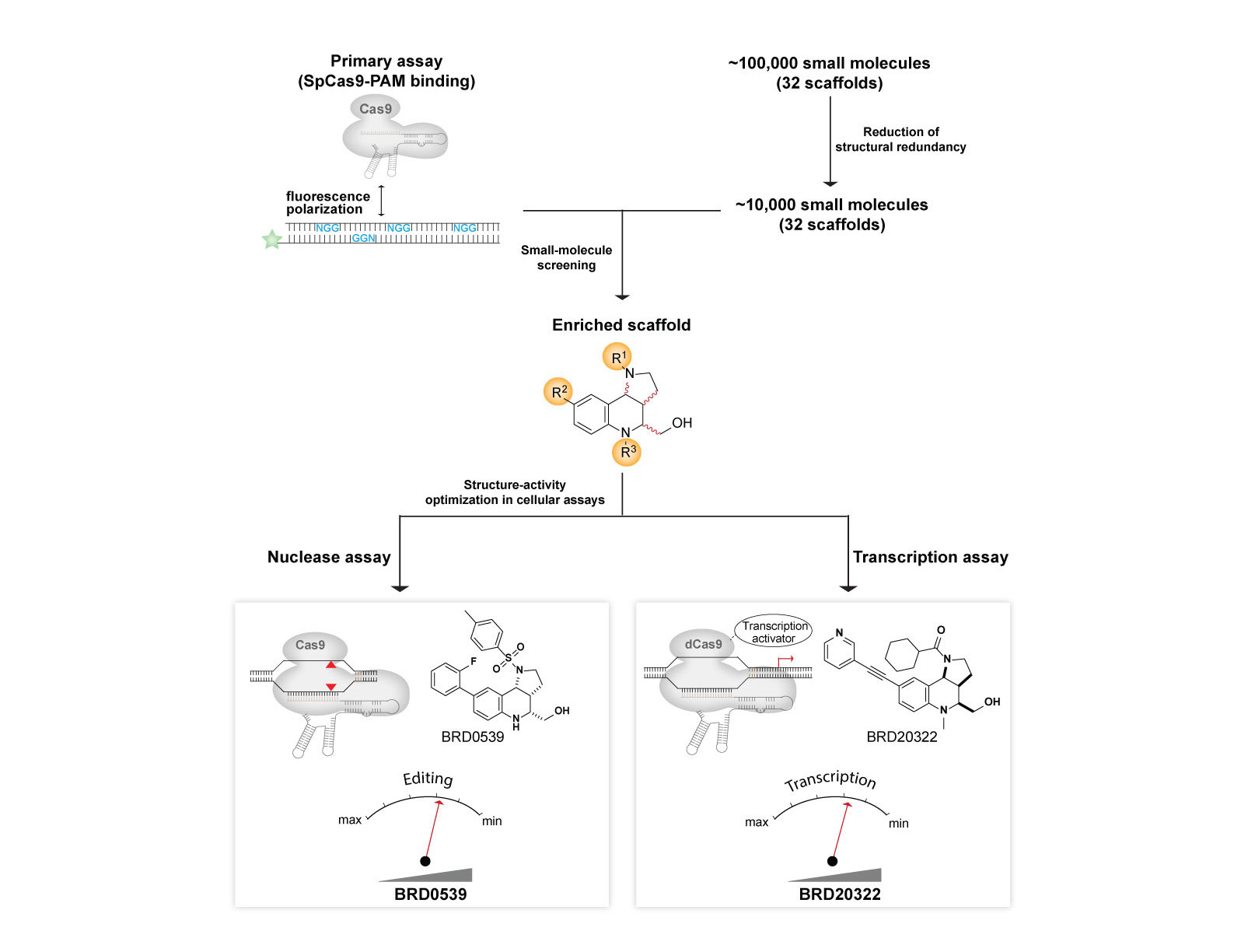
We created a generalizable platform that provided the first synthetic inhibitors of Cas9 that weigh <500 Da and are cell-permeable, reversible, and stable under physiological conditions. We developed a suite of high-throughput assays for Cas9 functions, including a primary screening assay for Cas9: DNA interaction, and used these assays to screen a structurally diverse collection of natural-product-like small molecules to ultimately identify inhibitors. Using these synthetic anti-CRISPRs small molecules, we demonstrated dose and temporal control of Cas9 and catalytically impaired Cas9-based technologies, including transcription activation.
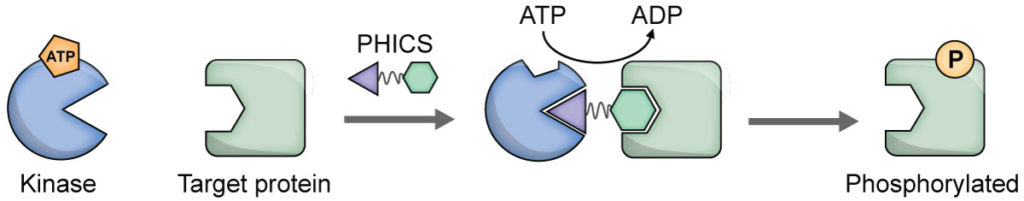
Several next-generation CRISPR-associated nucleases have emerged (e.g., SaCas9, Cas12a) with superior attributes. For example, SaCas9 is much smaller than SpCas9.19 While SpCas9 catalyzes blunt cuts, Cas12a leaves sticky ends. Cas12a employs smaller guide RNA and also can self-process an RNA sequence containing multiple gRNA target sequences into multiple gRNAs, thereby allowing easy multiplex genome editing. We have developed a suite of high-throughput assays for SpCas9, SaCas9, and Cas12a and performed small-molecule screening to identify inhibitors of these enzymes, including those that inhibit all the three enzymes (manuscripts in preparation)